تحديد المتغيرات الجينية النادرة المرتبطة بمرض الصمام الأورطي
تمكن باحثون من التعرف على المتغيرات الجينية المرتبطة بشكل نادر من مرض الصمام الأورطي ثنائي الشرفات الذي يصيب الشباب ويمكن أن يؤدي إلى مضاعفات أبهرية خطيرة ومهددة للحياة.
نشرت الدراسة في المجلة الأمريكية لعلم الوراثة البشرية.
وحسب ما نشره موقع ميديكال إكسبريس، قال الدكتور سيدهارث براكاش، الباحث الرئيسي المشارك في الدراسة والأستاذ المساعد في علم الوراثة الطبية وطب القلب والأوعية الدموية في قسم الطب الباطني في كلية ماكجفرن الطبية في جامعة تكساس للصحة في هيوستن: "لقد وجدنا سابقًا أن الأفراد الشباب الذين يعانون من تشريح الأبهر الصدري المبكر هم أكثر عرضة للإصابة بصمامات الأبهر ثنائية الشرفات وأكثر عرضة للإصابة بمتغيرات نادرة في الجينات المرتبطة بصمام الأبهر ثنائي الشرفات".
وأضاف: "عندما لاحظنا أن صمام الأبهر ثنائي الشرفات هو نوع من مؤشر الخطر لهذه المجموعة مع نتائج سيئة، أردنا على وجه التحديد معرفة ما إذا كان الأفراد الشباب الذين يعانون من مشاكل سريرية تتعلق بمرض صمام الأبهر ثنائي الشرفات قد يكون لديهم أيضًا متغيرات جينية نادرة تتنبأ بمضاعفات مثل الحاجة إلى جراحة الصمام".
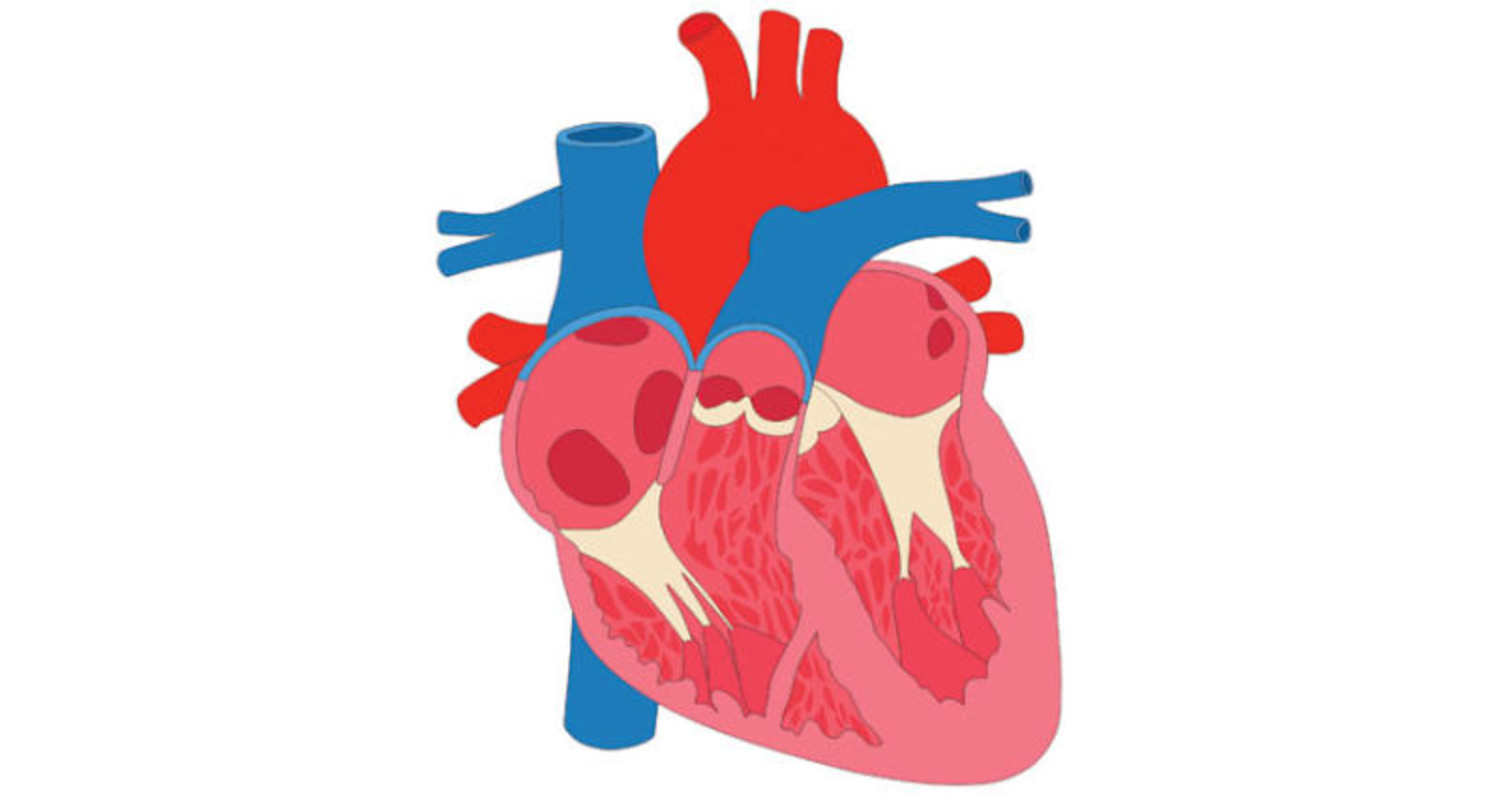
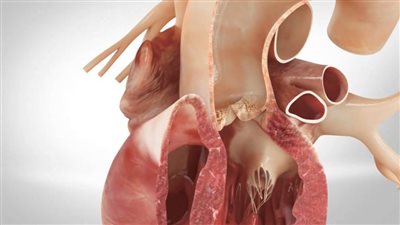
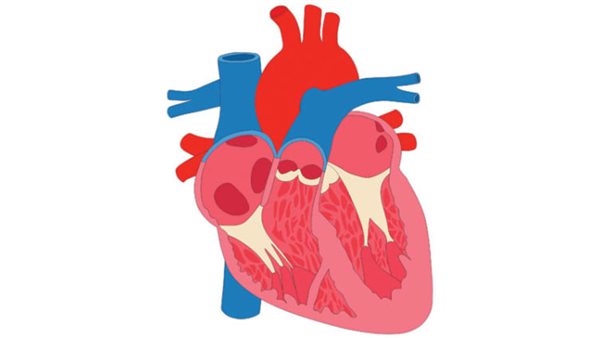
الصمام الأبهري
يولد حوالي 1 من كل 100 شخص بصمام أبهري ثنائي الشرفات، مما يجعله السبب الأكثر شيوعًا لأمراض القلب الخلقية.
سمحت المقارنة بين المجموعة الفرعية النادرة من مرضى الصمام الأورطي ثنائي الشرفات المبكر مع السكان الشائعين لهذا المرض للباحثين بتحديد المجموعة من المرضى التي ستستفيد من الاختبار الجيني، مما يتيح العلاج المبكر الأكثر قوة.
وبحسب الباحثين، فإن المرضى الذين يعانون من مرض الصمام الأورطي ثنائي الشرفات غالبًا ما ينتظرون وقتًا طويلًا حتى يتم فحصهم، مما يؤدي إلى أعراض قلبية وعائية أكثر شدة، مثل قصور القلب وحتى الموت المفاجئ.
صمام الأبهر ثنائي الشرفات هو عيب خلقي في القلب حيث يحتوي الصمام على رفرفين أو شرفتين بدلًا من ثلاث، وبالتالي لا يفتح الصمام ويغلق بشكل صحيح مع كل نبضة قلب.
يمكن أن يؤدي هذا إلى مضاعفات مثل انسداد أو انخفاض أو تدفق الدم للخلف عبر حجرات القلب، مما يسبب ضيق التنفس وألم الصدر والإغماء وصعوبة ممارسة الرياضة. في الحالات الأكثر شدة، يمكن أن يؤدي المرض إلى تشريح الأبهر، أو تمزق الشريان الأورطي، وهي حالة تهدد الحياة.
درس الباحثون الأفراد الذين يعانون من مضاعفات محددة للمرض قبل سن الثلاثين أو الذين كانوا أقارب مباشرين لشخص مصاب بمرض الصمام الأبهري ثنائي الشرفات المبكر.
تم تعريف أعراض المرض المبكرة على أنها تضيق أو ارتجاع أبهري متوسط أو شديد، أو تمدد الأوعية الدموية الأبهري الصدري الكبير، أو الحاجة إلى إجراء جراحة أبهرية، أو تشريح الأبهر.
كان هدف الباحثين هو تحديد المتغيرات الجينية التي قد تؤدي إلى زيادة خطر الإصابة بالمرض بين الشباب.
وقال براكاش: "كان الشخص العادي في هذه الدراسة مصابًا بالمرض في العشرينيات من عمره وكان لديه أقارب مصابون بالمرض، لذلك تتبعنا بداية المرض في العائلات وأبلغنا عن المتغيرات الجينية النادرة التي انفصلت مع المرض في هؤلاء المشاركين وأقاربهم".
قام براكاش وفريقه بتحليل بيانات تسلسل الإكسوم الكامل، والتي تم الحصول عليها من 215 عائلة من أكثر من 20 مؤسسة لتحديد المتغيرات الجينية النادرة المعروفة بأنها تسبب أمراض القلب الخلقية في مرض الصمام الأورطي ثنائي الشرفات المبكر في هذه المجموعة الفرعية النادرة، وقارنوا هذه النتائج بالسكان الأكثر شيوعًا من المرضى الذين يعانون من مرض الصمام الأورطي ثنائي الشرفات المتأخر.
تضمنت الجينات التي تم تحديدها جينات تسبب صمام الأبهر ثنائي الشرفات غير المتلازمي المعزول، فضلًا عن أنواع أخرى من أمراض القلب الخلقية المرتبطة بصمام الأبهر ثنائي الشرفات أو التشوهات الخلقية ذات الصلة. ووجد الباحثون متغيرات ضارة من الجينات مع أدلة معتدلة أو قوية على أنها تسبب أنماط القلب التنموية في 107، أو 50٪، من العائلات المتضررة في الدراسة.
وقال براكاش: "أظهرنا أن المرضى الأكبر سنا الذين لديهم صمامات أبهرية ثنائية الشرفات من غير المرجح أن يستفيدوا من الاختبارات الجينية لأن من غير المرجح أن يكون لديهم هذا النوع من المتغيرات الجينية".
وأضاف: "من المهم أن يدرك الناس، كما رأينا في هذه الدراسة، أن العديد من الأشخاص الذين يعانون من صمامات الأبهر ثنائية الشرفات لديهم أقارب مصابون. وفي المستقبل، قد يتم اختبار أفراد الأسرة بحثًا عن المتغيرات الجينية التي تسبب مضاعفات صمامات الأبهر ثنائية الشرفات، ويمكن علاج الأشخاص الذين لديهم هذه المتغيرات الجينية في وقت مبكر لمنع حدوث مضاعفات مستقبلية".